
Start the Clock
Preparation of a single rotational quantum state of NO(A2∑+) in a crossed molecular beam apparatus is used to investigate the stereodynamics of rotational energy transfer in encounters with a range of collision partners.
|
|
|

|
Laser preparation of a collision species precisely defines a zero-point in the time period over which collisions may occur, and the resulting images may be straightforwardly interpreted to resolve the detailed spatial properties of the collision process. |


Start the Clock: A New Direct Method to Study the Collisions of Electronically Excited Molecules
This EPSRC-funded project employs a crossed molecular beam velocity map ion imaging (CMB-VMI) apparatus to investigate the dynamics of rotationally inelastic collisions of electronically excited NO(A2∑+) in the gas phase. Owing to the relative difficulty of preparing electronically excited species for use in scattering experiments, their collision dynamics have not received anything like the same level of attention paid to ground-state species, despite the important role that collisional energy transfer for such highly excited species plays in high energy environments such as combustion systems and plasmas.
These experiments employ pulsed laser radiation to prepare NO(A) in the region of intersection between molecular beams containing NO and a collider gas, and to probe the NO(A) molecules using Resonance Enhanced Multiphoton Ionization after collisions have been allowed to occur. Velocity Map Imaging of the ionized molecules produces an image which is a 2D projection of the laboratory frame velocity distribution. The state preparation and probe steps are rotationally state selective, meaning that collision-induced transitions between individual pairs of rotational states can be examined in isolation. The well-defined start and finish times imposed by the pump and probe steps mean that the experiment directly measures the scattered flux, rather than just the instantaneous density. This in contrast to conventional CMB scattering experiments, where collisions occur throughout the molecular beam pulse, typically 100 microseconds or more. Differential loss of fast and slow moving scattered molecules in conventional experiments requires a tricky density to flux transformation in the data analysis, something that the 'Start the Clock' method avoids. This is apparent in the excellent degree of symmetry about the relative collision velocity displayed in our images.

|
Images acquired for NO(A)+Ne scattering, in real time. Each spot is the result of an single ion hitting the detector. The initial direction of the NO containing molecular beam is towards the centre bottom of the image, where the most intense scattering is observed. |
The resulting images may be analysed to extract the differential cross sections (DCSs), the distribution describing the direction into which the products scatter, relative to the initial collision velocity. The product rotational angular momentum may also be polarized, i.e. the products may be preferentially rotating in a particular plane, or even with a particular handedness, as a function of the scattering angle. By controlling the polarization of the probe laser beam and taking images with different polarizations relative to the scattering plane we can also determine this scattering angle-dependent product rotational angular momentum polarization. After initial experiments carried out in collaboration with Dr David Chandler of the Combustion Research Facility at Sandia National Laboratories, we designed, constructed and commissioned a new CMB-VMI apparatus at Heriot-Watt specifically for collisions of electronically excited molecules. We have performed a range of experiments exploring collisions with atoms and molecules, and have interpreted these results with quantum and classical scattering calculations on ab initio potentials.
Scattering from Ar and Ne atoms: Testing ab initio potential energy surfaces and observing non-rigid polarization dynamics
We have studied the stereodynamics of the collisions of NO(A) with Ar and Ne, the latter with two different collision energies. In each case the experiments were polarization sensitive, providing measurements of the product rotational angular momentum alignment as a function of the scattering angle, as well as the differential cross sections. We compared the experimental results to QS calculations on recent ab-initio potential energy surfaces. This provides a direct test of the ability of high level electronic structure theory to accurately predict the energies in the NO(A)-Ar and NO(A)-Ne systems. In general, we found good agreement between experiment and theory, but there are clear disagreements in some areas. For example, our measurements (and earlier proof-of-concept measurements made by our collaborators at Sandia National Lab) for the NO(A)+Ne system clearly show a forward scattered peak in the differential cross sections for a range of final N' states, which is not reproduced by theory. This is strong evidence for the presence of a (shallow) attractive well in the NO(A)-Ne PES that is not predicted by the ab initio potential calculations. The precision of our measurements enabled us to test two different recent potentials for the NO(A)-Ne system, and comment on their relative accuracies.
|
(Left) Colour map images for NO(A, v = 0, N = 0)) + Ne scattering to final state N' = 7, both experiment and fit to extract the differential cross section and polarization moments. The extracted DCS is shown in the upper panel (black), together with the result of quantum scattering calculations (blue). Note the extreme forward peak (0-10 degrees) not predicted by theory. (Right) Angle-resolved rotational alignment moment from the same measurement. The data (black) and quantum scattering calculations (blue) are in good agreement, but are very different to the predictions of the classical, hard-shell, kinematic apse model (red). |
-Ne_N7_DCS.png)
-Ne_N7_A20.png)
Another interesting and previously unknown feature of the dynamics of these systems is seen in the rotational angular momentum polarization of the products. The product polarization as a function of scattering angle has only been measured for a limited range of collision systems to date, e.g. NO(X) + rare gases and CO(X) + rare gases. In these systems, a simple classical model 'kinematic apse conservation' accurately predicts the observed dynamics, consistent with conservation of angular momentum in collisions with a rigid shell. However, in the NO(A) + rare gas systems studied here, the product rotational alignment shows strong deviations from this kinematic apse model, which are accurately predicted by quantum scattering theory. We currently believe that this is a consequence of the non-rigid nature of the NO(A)-Rg potential energy surfaces, but the question of whether it is also a purely quantum mechanical property is still open.
Scattering from He and D2: Comparing atomic and molecular colliders with identical kinematics
In this work, we studied the collisions with two different colliders with the same mass, He and D2. An accurate ab-initio PES exists for the NO(A)-He system, but (at the time) no such PES had been calculated for the NO(A)-H2/D2 system. By comparing the dynamics of the two systems at identical collision energies we hoped to be able to identify common elements and thereby make some predictions about the nature of this unknown NO(A)-H2 PES.
-He&D2_images-1.png)
-He&D2_DCS.png)
|
The agreement between experiment and theory for NO(A)-He is quantitative, indicating that the NO(A)-He PES is correct to the limits of the experimental test. The observed scattering for NO(A)+D2 is very similar in form to that for NO(A)+He, but the observed rainbow scattering peak is shifted to forward (smaller) angles for each measured N'. This is consistent with the NO(A)-D2 and NO(A)-He PESs having a very similar form with respect to NO(A), with a slightly more attractive (or more likely, slighly less repulsive) PES for D2.
In principal, the D2 could also be rotationally excited in the collision. This would result in scattering at slower centre-of-mass speeds, as some of the available energy is channeled into rotation instead of translation. We looked for evidence of this, which we would expect to be particularly apparent in the higher NO(A) N' which result from low impact parameter 'hard' collisions. However, we saw no evidence for any excitation of the D2. This suggests that the PES is relatively isotropic with respect to the D2 i.e. the D2 looks quite spherical to the approaching NO(A). This, coupled with the large rotational constant (ca. 30 cm-1) of D2, results in no rotational excitation of the D2 at this collision energy.
Scattering from N2: Pair-correlated differential cross sections
|
|
-N2_N=11_Newton.png)
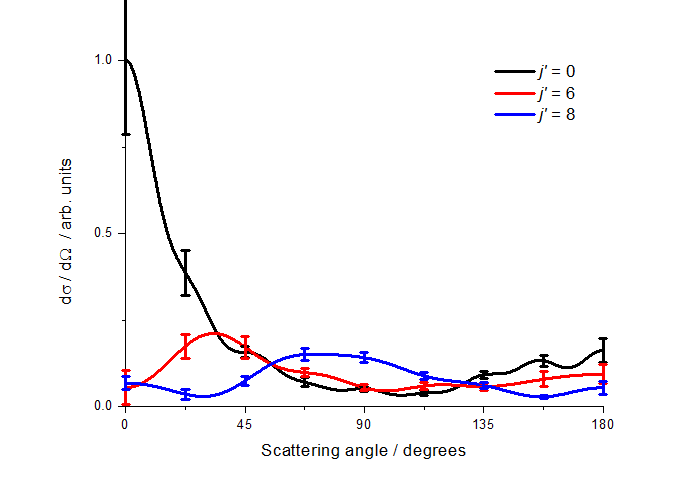
N2 has a rotational constant that is very similar to that of NO(A) (ca. 2 cm-1), and much smaller than that of D2 used in the previous study. We therefore made a systematic study of the collisions of NO(A) with N2, in which we fitted the experimental images to extract differential cross sections as a function of the internal (rotational) energy of the unobserved collision partner. This is possible because depositing energy in the N2 rotation means that there is less energy available for translation of the NO and N2. Scattering in which the collider has been rotationally excited therefore leads to additional intensity in the inner parts of an image. If the rotational spacing was larger than the collisional energy spread, this would lead to clear superimposed scattering images. However, in these experiments this is not the case, and the different scattering circles are blended together. This is illustrated in the figure, which shows the measured image for the NO(A) N' = 11 product state, overlaid with a Newton diagram. The different rings indicate increasing internal energy in the N2, namely 0, 84, 144, 220, 312 and 420 cm-1.
We fitted the experimental images simultaneously to three discrete N2 internal energies: 0, 84 and 144 cm-1, corresponding to j = 0, 6 and 8, which extensive simulation and testing suggested were sufficiently separated to be distinguishable. In this fashion we were able to extract the DCSs for correlated excitation of the NO(A) and the N2.
Relatively little rotational energy is transferred into the N2 in total, but there is a clear positive correlation with NO(A) rotational energy, i.e. as the NO(A) rotates faster, so does the N2. The DCSs for correlated rotational excitation are somewhat surprising, as the highest joint rotational excitation occurs with sideways scattering. As shown in the DCS figure, backwards scattered NO(A) N' = 11 is preferentially formed in coincidence with low rotational state N2. These are some of the first measurements of rotational-state correlated differential cross sections, which we hope will help to stimulate theoretical modelling of these molecule-molecule energy transfer processes.
Scattering from CO2: Pair-correlated differential cross sections
|
|
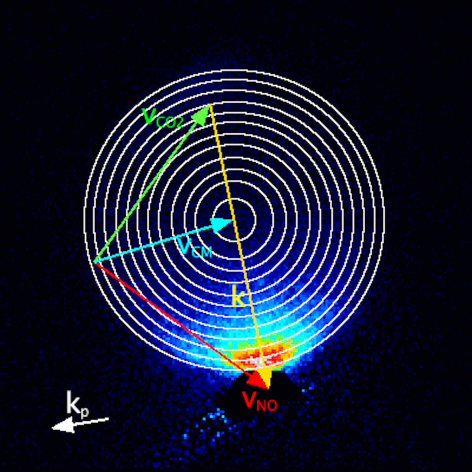
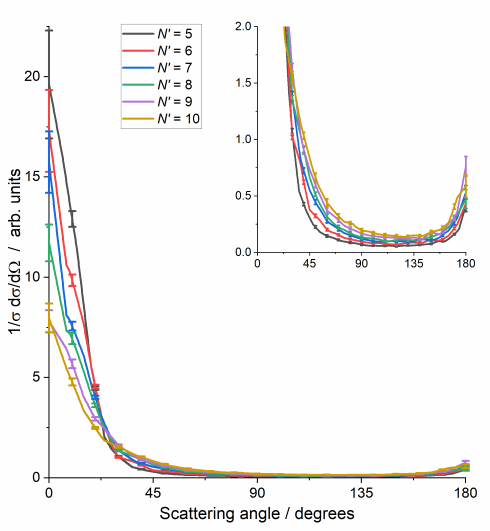
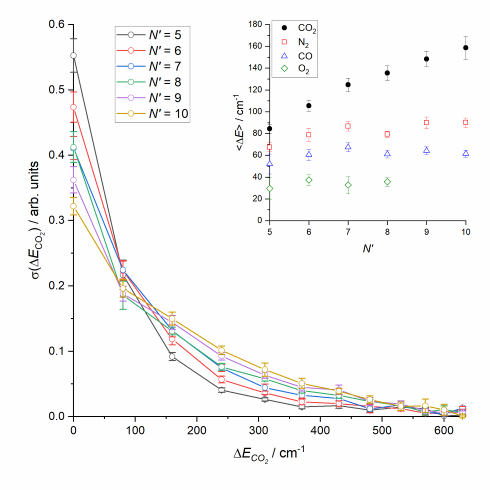
CO2 has a significantly smaller rotational constant than NO(A) (ca. 0.39 cm-1 rather than 2 cm-1). CO2 is also a very efficient quencher of NO(A), such that the majority of collisions will lead to inelastic or reactive removal of NO(A), rather than rotational energy transfer within the NO(A) state. We therefore made a systematic study of the collisions of NO(A) with CO2, in which we again fitted the experimental images to extract differential cross sections as a function of the internal (rotational) energy of the unobserved CO2.
The differential cross sections are all very strongly forward scattered, regardless of the final rotational state of the N2. A small backward scattered peak is seen, which increases in size for the higher NO N'. The surprising observation is that there is no significant sideways scattering, even for high rotational energy transfer to the NO. Simultaneously, there is significant coincident rotational excitation of the CO2, for all final rotational states of NO(A). This is very surprising, we can see strong rotational excitation of both products, but with forward scattering dominating in all cases. It is not consistent with interactions with a rigid repulsive wall.
|
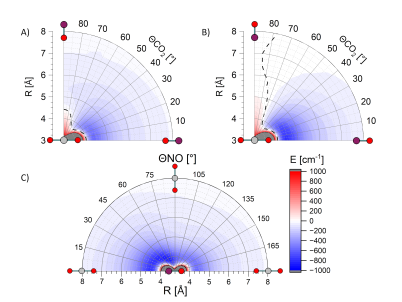
Potential energy surfaces were calculated by the Paterson group for the van der Waals region at the CCSD(T)-F12a level of theory. At short internuclear distances (R < 4 Angstrom) CCSD(T) becomes unreliable as the multireference chraracter of the system increases. We therefore can assume that close approach leads to quenching with high efficiency, consistent with the literature quenching kinetics for the system. Hence, there are essentially no inelastic repulsive wall collisions for this system, and low impact parameter collisions that would usually lead to sideways or backwards scattering with medium to high rotational excitation instead disappear down the 'black hole' of the quenching conical intersection. |
However, the van der Waals region of the PES is strongly attractive, and strongly anisotropic with respect to both the NO and the CO2. We believe that this is the origin of the strong rotational excitation of both fragments with forward scattering. The deep well at long-range for the linear-linear conformation of the collision system is able to provide the rotational torque required with small angular deflections. An alternative interpretation is that a transient collision complex is formed, mediated by the deep well, and that subsequent dissociation of the complex (an 'osculating complex') leads to the forward/backward DCS. The backward peak is also very similar to the 'backward soft collision glories' recently observed in NO(X)+NH3 scattering, a feature that requires a well depth comparable to the collision energy, which is the case in these NO(A)+CO2 collisions.
4-vector correlations: Rotational reorientation as a function of scattering angle
|
|
+Ne_Orientation.png)
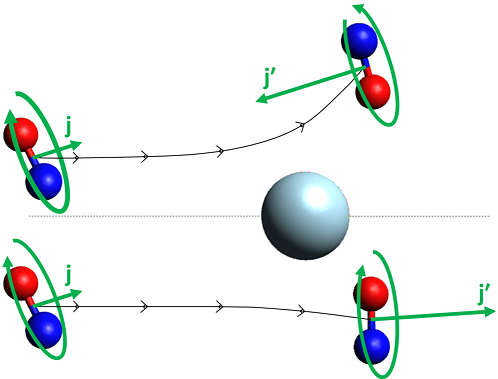
Optical state preparation provides an opportunity to vary the initial rotational state, something that is difficult to achieve with other state preparation methods (e.g. hexapole state selection). In preparing an initially rotating NO(A), we can also use the polarization of the excitation laser to prepare the molecules with an initial rotational orientation, i.e. with an preferred handedness of rotation relative to the collision velocity. In this experiment, we used circularly polarized excitation light to prepare the NO(A) N = 2, j = 1.5 state with an orientation of its rotational angular momentum pointing along the collision velocity. We then collided it with Ne, and probed the scattered products using circularly polarized probe light. In this fashion, we were able to measure the degree of retention of the orientation as a function of scattering angle for the products. This is sensitive to the full 4-vector correlation between initial and final translation and rotation, the k-j-k'-j' correlation, and this was the first time that such a direct measurement of a 4-vector correlation had been made in inelastic scattering.
The measured orientation, represented here by the phenomenological parameter C, varies dramatically with scattering angle and as a function of product state, N'. In general, strong conservation of the orientation is observed for extreme forwards and backwards scattering, consistent with conservation of angular momentum. However large deviations away from this are observed over small scattering angle ranges. For one state, N' = 5, we even observe a change in sign of C for a range of sideways scattered angles. In this range, the product NO is preferentially rotating with opposite handedness to the initially created polarization. A simple classical hard-shell (kinematic apse) model (blue points) does not capture any of this behaviour. In contrast, quantum scattering calculations do predict the main features, although we find that there is poorer agreement for this quantity with experiment than is observed for the more conventional vector correlations of the DCS etc. This is strong evidence that this more highly correlated measurement is more sensitive to the underlying potential energy surface than the more averaged 'conventional' vector measurements.
-
Inelastic Scattering of NO(A2Σ+) + CO2: Rotation-Rotation Pair-Correlated Differential Cross-Sections
Faraday Discussions (2024) 251, 279
doi: 10.1039/D3FD00162H
-
Differential Cross Sections for Pair-Correlated Rotational Energy Transfer in NO(A2Σ+) + N2, CO, and O2: Signatures of Quenching Dynamics
Journal of Physical Chemistry A (2023) 127, 6251
-
Stereodynamics of rotational energy transfer in NO(A2Σ+) + Kr collisions
Physical Chemistry Chemical Physics (2022) 24, 6525
doi: 10.1039/D1CP05960B
-
Non-intuitive rotational reorientation in collisions of NO(A2Σ+) with Ne from direct measurement of a four-vector correlation
Nature Chemistry (2018) 10, 1148
-
Pair-correlated stereodynamics for diatom-diatom rotational energy transfer: NO(A2Σ+) + N2
Journal of Chemical Physics (2017) 147, 013912
doi: 10.1063/1.4979487
-
Experimental testing of ab initio potential energy surfaces: Stereodynamics of NO(A2Σ+) + Ne inelastic scattering at multiple collision energies
Journal of Chemical Physics (2016) 145, 174304
doi: 10.1063/1.4966688
-
Comparative stereodynamics in molecule-atom and molecule-molecule rotational energy transfer: NO(A2Σ+) + He and D2
Journal of Chemical Physics (2016) 145, 084312
doi: 10.1063/1.4961258
-
Rotationally Inelastic Scattering of NO(A2Σ+) + Ar: Differential Cross Sections and Rotational Angular Momentum Polarization
Journal of Chemical Physics (2015) 143, 204301
doi: 10.1021/1.4935962
-
Rotational Alignment of NO (A2Σ+) from Collisions with Ne
Journal of Physical Chemistry A (2013), 117, 8163
doi: 10.1021/jp402019s
Collisions of Electronically Excited Molecules: differential cross-sections for rotationally inelastic scattering of NO(A2Σ+) with Ar and He
Molecular Physics (2012), 110, 1693
Direct angle-resolved measurements of collision dynamics with electronically excited molecules: NO(A2Σ+) + Ar
Journal of Chemical Physics (2011), 134, 091101
doi: 10.1063/1.3563016
Go to top